Fragment Screening & Hit Identification: Which Library to Choose?
In this third post on structural biology and structure-based drug design we discuss fragment libraries and fragment-based lead discovery
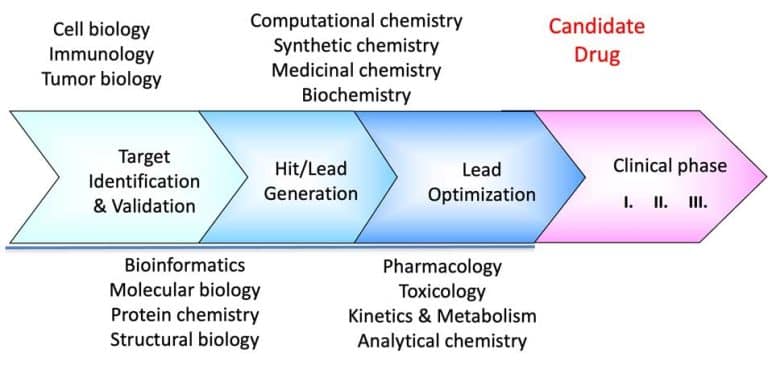
We continue our series on structural biology and structure-based drug discovery and will further discuss fragment screening and hit identification and the choice of a screening library. As I mentioned in the previous post, screening is central to any drug discovery strategy we may choose. Screening aims to identify a compound (hit identification) or a series of compounds that can bind to the drug target and modify its activity. Before the start, we need to define two essential factors: the type of library we want to use and the screening method.
How do I choose the library? We know that the chemical space is endless. However, the good news is that there is no need for a library with billions of compounds in your fragment screening and hit identification. We can limit the number of compounds for screening in many ways. One of the best-known is Lipinski’s rule of five (Lipinski et al., 2001) for orally available drug candidates. This is an empirical rule based on the analysis of known drug molecules. The rule states that an orally active drug should not have more than one violation of the following criteria:
Mw < 500 g/mol
Hydrogen bond acceptors < 10 (N and O)
hydrogen bond donors < 5 (OH and NH)
logP < 5 (logP=log ([Coctanol]/[Cwater])
logP is needed to ensure the molecules’ solubility is at a certain level. It describes the distribution of the compound in an octanol-water system, where [Coctanol] and [Cwater] are the concentrations of the compound in the octanol and the water phase, respectively. These conditions will limit the potential number of compounds in the library. We could further limit our choice by selecting compounds similar to known lead compounds (lead-like compounds) targeting a particular target class by defining shape complementarity to fit a specific binding site. Another essential factor is, of course, the chemistry of the compounds. They should be easy to modify chemically and should not contain atomic groups known to trigger a toxic response or unwanted reactions. Limiting the number of rotatable bonds (maximum seven) has also been an important parameter (Veber et al., 2002).
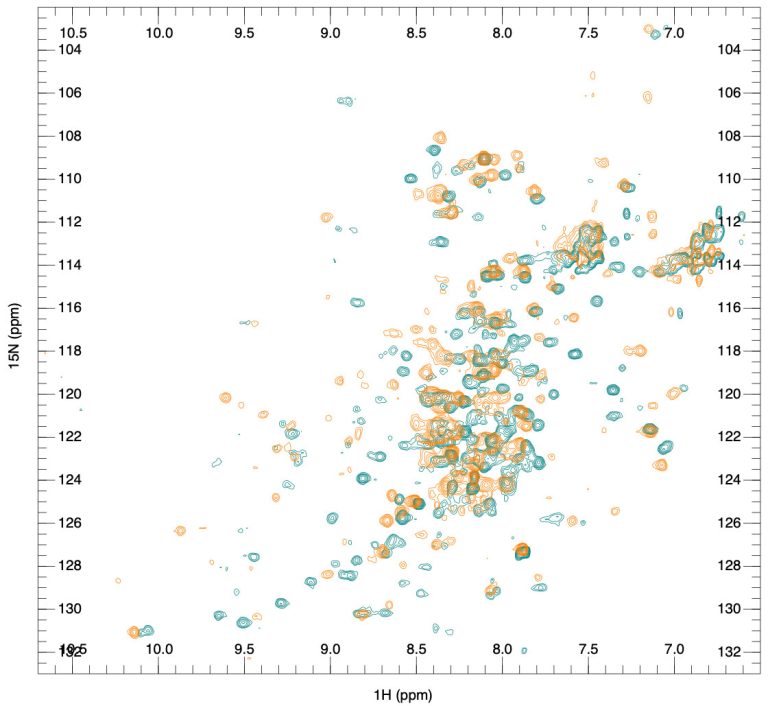
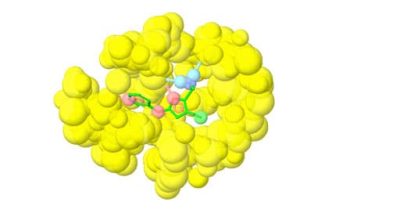
Fragment-based lead discovery is an alternative approach to drug-like or lead-like compounds, which has gained considerable popularity due to its efficiency in identifying a pharmacophore. A pharmacophore describes the interactions that contribute to ligand binding and can be used to design new compounds during hit-to-lead optimization and construct a screening library. Fragment screening uses a relatively small library with compound molecular weight in the 100-300 Da range. The small size of the fragments will minimize the chances of unfavorable interactions like steric repulsion, increasing the probability of binding to the target protein and the hit rate. This will identify weakly potent and biologically active molecules. Complexes of these molecules with the target can be studied with X-ray crystallography or NMR spectroscopy. They will guide the hit-to-lead expansion and optimization of affinities and specificities into potent leads during hit-to-lead optimization.
A fragment library can be constructed to sample a much larger chemical space than lead-like or drug-like libraries. Another popular approach involves the creation of DNA-encoded libraries (DELs). DELs have the advantage of allowing an even larger chemical space coverage (for details, see, e.g., this summary and some reference publications).
For constructing a fragment library, the rule of three has been suggested (Congreve et al., 2003):
MW 100-300
LogP ≤ 3.0
H-Bond Acceptors ≤ 3
H-Bond Donors ≤ 3
Rotatable bonds ≤ 3
Polar Surface Area ≤ 60 Å2
X-ray crystallography and NMR spectroscopy can be used for fragment screening, hit identification, and studying compound binding. Generally, in fragment screening, biophysical methods quickly became popular since they provide a rapid and efficient assessment of the binding of weak hits, which can be challenging to identify in biochemical assays. Other methods used in fragment screening include:
Surface plasmon resonance (SPR)
Thermal shift assay (differential scanning fluorimetry, DSF)
Weak affinity chromatography (WAC™)
SARomics Biostructures’ structural biology services include fragment screening and hit identification services using X-ray crystallographic and NMR spectroscopy screening, as well as the WAC™ screening techniques. WAC™ is a proprietary method jointly owned by SARomics Biostructures and our partner, Red Glead Discovery. The method uses a chromatography column to inject a solution of fragments or lead-like compounds. During elution, the fragments with a higher affinity for the protein will stay on the column longer than those with a low affinity. Mass-spectrometry can conveniently detect and identify the fragments, thus establishing a direct ranking of hits. WAC™ is an efficient, high-throughput, and lower-cost choice compared to other biophysical screening methods. In addition, we offer our proprietary library of MedChem-friendly low-molecular-weight fragments designed to be general-purpose (not target-directed), covering diverse chemical space. Of course, customers’ libraries may also be used for screening. Several successful projects have already demonstrated the efficiency of the method.
We will continue this series of posts on structural biology and structure-based lead discovery and design. The following post will discuss the strategies for structure-based drug design. Follow us on LinkedIn to ensure you do not miss our future posts!
See also structure-based drug design strategies page in our website’s methods and knowledge section.
Our drug discovery services can be found in fragment screening & integrated drug discovery.